
БНБ
"БРОКГАУЗ И ЕФРОН" (121188)
- Photogallery
- Естественные науки - Математика - Технология
- Авиация и машиностроение
- Высокие технологии
- Вычислительная техника
- Нанотехнология
- Роботехника
- Энергетика
- Электроника
Электрохимия
Определение "Электрохимия" в словаре Брокгауза и Ефрона
Электрохимия
Электрохимия*
Содержание: Введение. Историческая справка. — Обозначения, принятые в Э. — Основные законы и принципы. — Перенос ионов. — Электропроводность растворов. — Электровозбудительная сила. — Переход химической энергии в электрическую. — Классификация гальванических элементов. — Электролиз. — Современные электрохимичесие теории.
Предмет Э. составляет изучение явлений, сопровождающих непосредственный переход химической энергии в электрическую и электрической в химическую. Химическая энергия среди других форм энергии играет роль собирателя богатства природы. В этой форме накоплены громадные запасы энергии; каменный уголь, торф, нефть, дерево и т. п. так называемое топливо, с одной стороны, и кислород воздуха, с другой. Непосредственный переход химической энергии топлива в другие формы энергии, кроме тепловой, совершается только при исключительных, труднодоступных условиях. В противоположность тому электрическая энергия в высшей степени удобна для использования. Электрическая энергия переходит сравнительно легко в другие формы энергии. Она является благодетелем, который везде и всюду, где появляется, оказывает неоценимые услуги человечеству. Становится теперь очевидным громадный теоретический и практический интерес, сосредоточенный на вопросе перехода химической энергии в электрическую, как на вопросе о рациональному использования богатств природы. В настоящий момент переход этот достигается сложным, окольным путем. Сначала сжигают топливо, т. е. переводят его химическую энергию в тепловую, в паровой машине тепловую энергию переводят в механическую и, наконец, в динамо-машине механическую энергию переводят в электрическую. При этом сложном пути теряется от 85 % до 90 % первоначальной химической энергии и, в лучшем случае, только 15 % её удается перевести в электрическую. Возможен ли в настоящее время иной путь? Возможно ли непосредственно в своеобразном гальваническом элементе из энергии топлива получить электрическую энергию? Современное положение этого вопроса выясняет отдел Э., трактующий о переходе химической энергии в электрическую. К этому же отделу относится общее изучение гальванических первичных элементов. Учение об этих элементах теснейшим образом связано с вопросом о переходе химической энергии топлива в электрическую. Теоретическая сторона этого учения выяснила общий вопрос: в каком случае химическое превращение может служить для получения гальванического тока. Неприменимость же в фабрично-заводской практике известных теперь гальванических элементов объясняется как раз тем обстоятельством, что они составляют также звено в цепи химическая энергия топлива — электрическая энергия. Ведь все материалы гальванических элементов получаются применением того же топлива: цинк элемента Даниеля, Бунзена, Грене, Лекланше и т. д. из своих окислов восстанавливается тем же углем, кислоты и т. п. материалы гальванических элементов получаются при участии того же топлива. Приблизительный подсчет показывает, что стоимость электрической энергии, добываемой гальваническими элементами, в среднем в сто раз превосходит стоимость той же энергии, добываемой динамо-машиной. Можно сказать, что путь от химической энергии к электрической, ведущий через динамо-машину, теперь еще выгоднее и короче пути, ведущего через гальванические элементы. Решение вопроса об элементе, в котором уголь или какое-либо иное топливо служило источником энергии, как это будет выяснено ниже, находится еще только в зачаточном состоянии; общий вопрос о практически выгодном гальваническом элементе не решен. Таким образом, в этом отделе электрохимии открыто широкое поле для будущих изобретений и открытий, и знакомство с теоретической Э. предохранит искателя в этой области от заведомо ложных щагов. Не меньший горизонт для исследователя и изобретателя открывается во второй области Э., т. е. в отделе её, трактующем о переходе электрической энергии в химическую. Здесь создаются не только совершенно новые производства продуктов, раньше с трудом добывавшихся в небольших количествах в лабораториях, как, например, производство карбидов, но и многие старые химические производства преобразуются в электрохимические. Достаточно упомянуть, что мировое производство алюминия химическим путем в 1885 г. равнялось 15 тоннам, современное же электрохимическое 1900 года составляет 6000 тонн. Химическое производство могло понизить цену алюминия до 20 франков за килогр., т. е. цена его была приблизительно в пятнадцать раз дороже меди, электрохимическое производство понизило стоимость алюминия до 3 франков за килограмм, т. е., хотя по весу алюминий и дороже меди, но объем алюминия одинаковый объему меди стоит теперь дешевле. Необыкновенная чистота осаждаемых током металлов, значительно повышающая их цену, развила особую промышленность: рафинирование металлов, т. е. их очистку путем электролиза. В 1901 г. в Америке было рафинировано 314000 тонн меди и в Европе 172000 тонн. Легкость регулировки производства, связанная с компактностью электрохимических приборов, играет также большую роль при переходе от химических производств к электрохимическим. Получение водорода и кислорода для горнов, получение веществ для беления тканей, воска и др. разработано в такой форме производства, что в любой момент, замыкая ток, можно получить количество, необходимое для данных целей. Для всей этой области электрохимии имеет решающее значение дешевый источник электрической энергии. Стремятся заменить получение электрической энергии из топлива иными источниками энергии. Их нашли в применении гидравлических сил природы: водопадов, рек и т. п.; их должно искать в утилизации других сил: силы ветра, силы приливов и отливов, даже, быть может, в силе современных наводнений. Электрохимические производства во многих случаях настолько выгодны, что, за отсутствием более дешевых источников энергии, пользуются все тем же окольным путем добывания её из топлива. Так, в Англии и Германии в 1900 г. около 60 % всей энергии, тратившейся в электрохимических производствах, добывалось из топлива. Своеобразное место в электрохимии занимают аккумуляторы, т. е. приборы, служащие для накопления химической энергии в такой форме, которая непосредственно переходила бы в электрическую энергию. Технические задачи при устройстве этих приборов имеют решающее значение. Теория их сводится к теории гальванического элемента при разряде аккумулятора и к теории электролиза при заряжении аккумулятора (см. Аккумуляторы).
Историческая справка. Первые проявления электрической энергии были открыты и наблюдались в виде действия статического электричества. Характерным отличием явлений статического электричества должно считать ничтожное количество электричества, участвующее в явлении, при громадном его напряжении. Химическое же превращение, чтобы стать заметным, требует, как раз наоборот, очень больших количеств электричества и незначительных напряжение. Оствальд в монографии "Elektrochemie, ihre Geschichte und Lehre" пишет: "Те ничтожные количества электричества, которые давали прежние несовершенные машины, были недостаточны, чтобы вызвать какие-либо бросающиеся в глаза (химические) явления. Мы видим, что физики столетия пройзводят всевозможные электрические эксперименты, меж тем химические явления, их сопровождающие, остаются незамеченными". Первые точные указания об изменении химического состава вещества при электрическом разряде принадлежат, по-видимому, Петру Бекария. Он заметил выделение меди при прохождении искры между кусками окиси меди. Эти наблюдения должны быть отнесены к середине XVIII в. Вслед затем Ван-Марум произвел целый ряд исследований над окислением металлов как в воздухе, так и в воде при действии электрической искры. В 1789 г. Ван-Труствик и Дейман потоком искр разложили воду. Характерное свойство электрической искры составляет значительно повышенная температура. Эта высокая температура вызывает при прохождении искры целый ряд химических превращений, совершающихся помимо каких-либо электрических сил, одним только повышением температуры, как напр., соединение водорода с кислородом и т. п. Такие превращения не составляют предмета изучения электрохимии. Однако существует ряд превращений, стоящих как бы на границе чисто теплового действия электрической искры и электролитического её действия. Для этих превращений мы не можем с уверенностью сказать, чтобы они вызывались одним термическим действием искры и чтобы при этом искра не играла специфической роли катализатора, т. е. ускорителя превращения. Такие превращения обыкновенно не совершаются только при повышении температуры в тех же размерах, что и при действии электрической искры. Одно из таких превращений сделалось популярным в последнее время. Открытое Пристлеем приблизительно около 1775 г., оно вскоре было подробно изучено Кавендишем. Кавендиш, пропуская продолжительное время (около месяца) поток искр через воздух, собранный над едким кали, доказал образование азотной и азотистой кислоты из воздуха. Это превращение не привлекало внимания исследователей со времени Кавендиша чуть ли не до наших дней. В самое последнее время оно сделалось предметом многих исследований и послужило темой многих патентов. Особенно выдвинулся вопрос о горении азота, т. е. соединении его с кислородом воздуха после опубликования работ лорда Рэлэ и Рамзая над выделением аргона из воздуха предварительным сжиганием азота воздуха потоком электрических искр. В 1902 г. образовалось в Америке общество (The atmospheric products Сº) с основным капиталом в миллион долларов, устроившее завод для получения горением атмосферных газов азотной кислоты. Завод этого общества находится в знаменитом районе электрохимических заводов, приводимых в действие силой Ниагарского водопада. История Э., составляющей главную часть современной Э., т. е. получение и действие гальванического тока началась, очевидно, с 1799 г., т. е. с того года, когда Вольта открыл свой вольтов столб — первую гальваническую батарею. Уже весной 1800 года Карлейль и Никольсон заметили выделение газов при прохождении гальванического тока через каплю воды. В сентябре 1800 г. Рихтер собрал уже отдельно кислород и водород. В том же году Дэви начал ряд своих знаменитых работ в области Э. Он изгнал из химии фантастическое предположение о том, что будто электричество превращает воду в кислоты и щелочи, и показал, что кислоты и щелочи, образующиеся при электролизе воды, — продукт примесей, загрязняющих в ничтожном количестве перегнанную воду. Примеси эти попадают в перегнанную воду, главным образом, благодаря незначительной растворимости стекла. Дэви доказал правоту своего взгляда, произведя электролиз в золотом сосуде. Дэви же принадлежит разложение электролизом расплавленного едкого кали и натра, т. е. выделение новых металлов калия и натрия из веществ, считавшихся до него простыми телами. Обобщив действие гальванического тока на химические соединения, Дэви дал первую электрохимическую теорию. Его теория представляет полное отождествление химических и электрических сил. Она связана логически с теорией контакта Вольты, согласно которой одного прикосновения разнородных веществ достаточно, чтобы вызвать на них противоположный электрический заряд. "Почему же не может быть" — пишет Дэви — "что электричество и сродство представляют одно и то же"... (цитировано по Каблукову). Итак, по Дэви, при соприкосновении частиц разнородных веществ они заряжаются противоположными электричествами и потом соответственно взаимно притягиваются, образуя таким образом химическое соединение. Открытое Дэви разложение считавшихся до него простых тел, произвело значительное впечатление на умы современников и весь период деятельности Дэви можно характеризовать как период выяснения вопроса: какие же вещества должно считать простым телом и какие сложным. "До работ о Дэви", — пишет Каблуков, — "едкие щелочи считались элементами, а хлор — сложным телом; после него щелочи оказались сложными телами, а хлор — элементом". Электрохимическая теория Дэви вскоре была вытеснена теорией Берцелиуса. По теории Берцелиуса все сложные вещества построены из двух частей; каждая из этих частей заранее обладает двумя электрическими полюсами. Сила полюсов у одного и того же атома или группы атомов не одна и та же, y металлов преобладают положительные полюсы, у металлоидов — отрицательные и т. п. Химическое соединение происходит взаимодействием сильнейших полюсов, скажем, при образовании повареной соли положительный полюс натрия взаимодействует с отрицательным хлора. Теория Берцелиуса оказала громадные услуги: современниками она дала толчок к систематическому изучению химических соединений и легла в основу их классификации. Эта теория обладала одним существенным недостатком: в ней еще смешивались два понятия — количество электричества и электрическое напряжение, и поэтому она пала под ударами открытий Фарадея. Она сделалась жертвой того закона Фарадея, который лег в основу современных электрохимических теорий. По теории Берцелиуса сила электрохимического сродства определялась количеством электричества на каждом полюсе, отделяющимся при электролизе группы атомов (ионе). Закон же Фарадея состоял именно в том, что одно и то же количество электричества нужно для разложения эквивалентных количеств разных химических соединений, т. е. для разделения их ионов. Берцелиус не мог примирить своей теории с законом Фарадея, оказавшимся одним из наиболее точных законов современной физики и химии. В настоящее время, конечно, мы ясно себе представляем, что хотя и одно и то же количество электричества нужно для разложения эквивалентных количеств любых веществ, но не одна и та же электродвижущая сила, а так как энергия измеряется произведением из количества электричества на электродвижущую силу, то и не одно и то же количество энергии. Фарадею мы обязаны и правильной, т. е. принятой в настоящее время, интерпретацией явлений, наблюдаемых в гальваническом элементе. Начиная с Вольты и чуть ли не до наших дней, по крайней мере до начала восьмидесятых годов, шел непрерывный спор о месте образования электричества в гальваническом элементе. Приверженцы классической теории контакта, принадлежащей самому Вольте, местом разделения положительного и отрицательного электричеств считали место соприкосновения металлов. Противники признавали, что разделение электричеств происходит там, где совершается химическое превращение. Фарадей был приверженцем химических толкований и, возражая приверженцам контактной теории, писал: "Это было бы по истине сотворением силы из ничего, подобного чему нет в природе". Этой фразой можно закончить историю развития учения о гальваническом элементе. Она была написана до формулировки закона сохранения энергии. Дальнейшее в учении о гальваническом элементе связано непосредственно с законом сохранения энергии и изложено ниже. Должно прибавить, что новейшие теории ясно рисуют картину происхождения гальванического тока, но самым существенным успехом должно считать: умение рассчитать соотношения между химической энергией и электрической. Вот это обстоятельство как бы предвидел Фарадей, писавший в 1838 году: "со временем мы будем в состоянии сравнить такие силы в телах, как тяжесть, сцепление, электричество, химическое сродство, и тем или иным способом выводить их относительные эквиваленты из их действий, теперь же мы этого не можем" (перевод Каблукова).
Обозначения, принятые в электрохимии. Для удобства дальнейшего изложения необходимо указать на обозначения величин, принятые в Э. Обозначения эти выработаны немецким электрохимическим обществом (Bunsen Gesellschaft) и доложены V-му международному съезду по прикладной химии в 1903 г. в Берлине.
| p, P | Обыкновенное и осмотическое давление |
| v | Объем |
| Θ | Температура по Цельзию |
| Т | Абсолютная температура Т = 273 + Θ |
| t | Время |
| Q | Количество теплоты |
| χ | Удельная электропроводность |
| η | Концентрация грамм-эквивалент в кубическом сантиметре |
| Λ | Эквивалент. электропроводность Λ = χ / η |
| Λ ∞ | Та же величина при бесконечном разбавлении |
| α | Степень электролитической диссоциации |
| Сохраняем здесь прежнее обозначение Аррениуса вместо γ — новое обозначение | |
| Электродвижущая сила в вольтах | |
| Е | Сопротивление в омах |
| W | Сила тока в амперах |
| J | Потенциал выделения, отдельный потенциал |
| ε | Потенциал выделения по отношению к водородному электроду |
| ε h | Потенциал по отношению к каломельному электроду |
| ε c | Механический эквивалент теплоты, равный 41,98.10 6 эрг для грамм-калорий при 15° (41,8.10 6 по новейшим определениям) |
| А | |
| Электрический заряд — эквивалент 96540 кулон (новейшие измерения дают 96580). | |
| F |
Основные законы и принципы. Электрохимия составляет часть физической химии, сравнительно молодой дисциплины, лежащей на границе физики и химии. Очевидно, основные законы и принципы электрохимии заимствованы, главным образом, от старших её дисциплин — физики и химии. Некоторые основные законы разрабатывались в последнее время исключительно в связи с электрохимическими задачами и имеют такое доминирующее значение для электрохимии, что о них должно быть здесь упомянуто. Первенствующий закон в электрохимии — это закон Фарадея; вкратце его можно формулировать: число разложенных током грамм-эквивалентов пропорционально количеству прошедшего электричества, причем фактор пропорциональности один для всех веществ. Под числом грамм-эквивалентов подразумевается вес выделившегося вещества, деленный на его эквивалент (см. Электрохимич. эквивалент). Очевидно, что при определении весовых количеств веществ, выделяемых на электродах при прохождении одного кулона электричества, должны получиться величины, пропорциональный эквивалентному весу данного вещества. Особенно тщательно определены количества серебра, выделяющиеся при разложении растворов азотнокислого серебра. Эти определения сделаны выдающимися исследователями и дали следующие результаты:
| миллиграммов | |
| Маскар | 1,1156 |
| Лорд Рэлэ | 1,1179 |
| Ф. и В. Кольрауш | 1,1183 |
| Потье и Пела | 1,1192 |
| Рихардс (1901 г.) | 1,1172 |
| Рихардс и Гемрод (1902 г.) | 1,1175 |
До определения Рихардса общепринято было, что кулон электричества выделяет 0,001118 г серебра, число, близкое к среднему из определений Рэлэ и Кольраушей. Очевидно, что количество электричества, нужное для выделения грамм-эквивалента серебра, т. е. 107,93 г, а следовательно, по закону Фарадея, и любого иного грамм-эквивалента, получится делением 107,93 на 0,001118. Это число обозначают F и до появления исследований Рихардса было общепризнанно его считать равным 107,93:0,001118, т. е. 96540. Новейшие исследования Рихардса и его учеников внесли небольшую поправку. Они были произведены с вольтметром (фиг. 1), в котором была введена диафрагма (а, фиг. 1) из пористой глины, защищавшая катод (с), платиновый тигель, от скорой диффузии к нему продуктов, образующихся в ничтожном количестве у серебряной палочки анода (b).
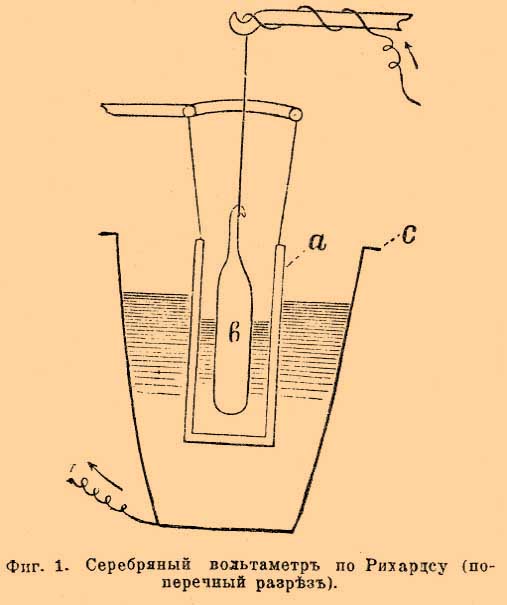
Фиг. 1. Серебряный вольтаметр по Рихардсу (поперечный разрез)
Раствором служит по-прежнему 15 % азотнокислое серебро. При этих условиях Рихардс окончательно установил вышеприведенное число 1,1175 мг и F = 96580. Число F, т. е. 96540 или 96580, смотря по тому, сохранить ли прежнее значение F или отдать преимущество новым определениям Рихардса, и будет характерным фактором пропорциональности закона Фарадея. Если через H обозначим эквивалентный вес и через n количество прошедшего электричества, через х весовое количество разложенного током вещества, тогда закон Фарадея можно выразить Fx/H = n, при постоянстве гальванического тока n = J t (см. обозначения) и, следовательно, x/H = J t/F. В электрохимии часто применяются первый и второй принцип термодинамики. Согласно первому принципу, т. е. принципу постоянства и эквивалентности энергий, электрическая энергия измеряется произведением количества электричества на разницу потенциалов, т. е. на величину электродвижущей силы, и равна EJt (см. обозначения) джоулей. Эквивалентность электрической и тепловой энергии определяется тем, что калория равна 4,18 джоулей, джоуль же 0,239 калории (мы принимаем за вероятное число для механического эквивалента теплоты 418.10 7 эрг или 42600 грамм-сантиметров). Очевидно, количество теплоты, выделяемое током в t секунд, равно 0,2392 E J t калорий. В электрохимии часто применяются законы Ома, Кирхгофа, Джоуля и др.; они изложены в статьях, трактующих об Электричестве и Гальванизме. Пользуясь обозначениями принятыми в электрохимии, закон Ома пишется J = E/W; закон Джоуля для количества теплоты, выделяемой током в секунду Q = 0,239 J2W калорий.
Прохождение тока через растворы электролитов. Явления, изучаемые в этом отделе, как по теоретическим причинам, так и по внешним признакам не могут быть отнесены только к случаям перехода электрической энергии в химическую. Возможно наблюдать прохождение очень слабых токов помимо разложения вещества током; при этом электрическая энергия будет переходить только в тепловую энергию (см. Электролиз). В самом же общем случае, изучение явлений при прохождении тока через растворы и сплавы электролитов (те же растворы только при высокой температуре) распадается на три части: во-первых, на изучение изменения концентраций растворенных веществ у электродов, т. е. изучение явления переноса ионов, во-вторых, на изучение электропроводности растворов и, в-третьих, на изучение продуктов, выделяющихся у электродов (электролиз). Современная теоретическая электрохимия не только качественно разъясняет происходящие явления, но и устанавливает количественные зависимости между величинами, наблюдаемыми при описанных явлениях. Выше приведенные законы (Фарадея, Ома и Джоуля) и принципы (сохранение энергии) служат основанием при нахождении большинства количественных зависимостей.
Перенос ионов. Рассмотрим случай, когда при прохождения тока часть растворенного электролита разлагается, и предположим для простоты, что разложившиеся части нацело выделяются из раствора. Опыты показали, что это разложение происходит отнюдь не равномерно во всем растворе. Все изменения раствора однородного вещества начинаются у обоих электродов и, по мере прохождения тока, распространяются от электродов к средним частям раствора. Средние же части раствора остаются совершенно не измененными, если, конечно, предохранить раствор от механического перемешивания выделяющимися у электродов газами или падением внутри раствора отделившихся от катода кусочков металла или какими-либо другими факторами. Опыт показала также, что изменение концентрации при разложении, скажем, ста частей какого-либо вещества отнюдь не совершается так, чтобы половина, т. е. 50 % его исчезло у одного электрода, а другие 50 % исчезли у другого. При электролизе разбавленного раствора йодистоводородной кислоты, из ста частей разложившегося йодистого водорода 17 % исчезают у катода и 83 % у анода; водород, конечно, выделится нацело у катода, а йод у анода. Попытки выяснить механизм выделения анионов и катионов у разных электродов нужно отнести еще к 1805 г. Гротгус (уроженец теперешних прибалтийских губерний) объяснил это явление, предположив, что при прохождения тока в растворе все молекулы располагаются так, что положительные их части, катионы по Фарадею, обращены к отрицательному полюсу, а отрицательные, анионы, к положительному и что при прохождении тока происходит обмен положительных ионов с ближайшими отрицательными; благодаря этому, у положительного электрода выделяется отрицательная часть молекулы, а у отрицательного положительная. Эта теория была усовершенствована маститым Гитторфом [Празднует в марте 1904 года восьмидесятый год от рождения] и доведена до современного её состояния 1853—1859 года. Гитторф разъяснял, что при таком двойном обмене должно происходить движение ионов — перенос ионов, причем анионы движутся к аноду, а катионы к катоду. Он же указал, что нет надобности считать, что оба вида ионов движутся с одинаковой скоростью для того, чтобы сохранилось неизмененное состояние раствора в неприлегающих к электродам слоях — в среднем слое у (см. фиг. 3). Фиг. 2-я представляет знаменитую молекулярную схему, предложенную Гитторфом.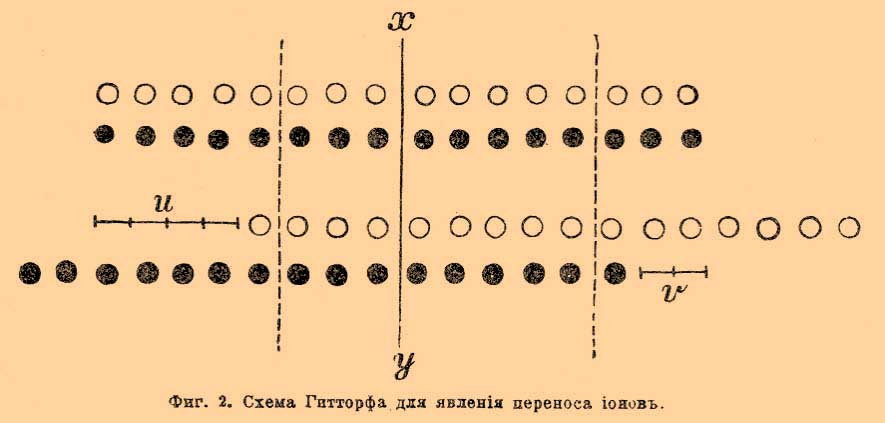
Фиг. 2. Схема Гитторфа для явления переноса ионов
Верхний двойной ряд кружков изображает состояние растворенных молекул до начала прохождения тока, нижний, — когда шесть молекул разложилось. Ясно, что число разложенных молекул по правую и левую сторону линии ху не одинаково, потому что белые кружки — ионы двигались в два раза скорее черных. Вернемся для фиксации понятий на частном примере к йодистому водороду. Легко показать, что приносимые током к электродам йод и водород, концентрации йодистого водорода менять не будут, так как нацело выделятся у электродов; будут же уменьшать концентрацию только уходящие от электрода ионы (см. фиг. 2), ибо благодаря их уходу и будет происходить разложение. Следовательно, уменьшение концентрации у каждого электрода будет происходить в зависимости от скорости уходящего от электрода йона. Если общее количество разложенного током вещества обозначим через m и через а количество вещества, исчезнувшего у анода, тогда a/m = n будет число, пропорциональное скорости переноса катиона (U). У катода исчезнет m—а вещества и (m— а)/m = 1 — а/m будет число пропорциональное скорости переноса аниона V. В случае йодисто-водородной кислоты, согласно выше приведенным данным для H· катиона, п равно 0,83, а для J' аниона 1—n равно 0,17. При обыкновенных опытах трудно подобрать условия, подобные электролизу йодистоводородной кислоты между угольными или платиновыми электродами, когда и водород, и йод не действуют на электроды. Обыкновенно изучают разложение солей между электродами из металлов, входящих в состав изучаемой соли, например раствор азотнокислого серебра между серебряными электродами, тогда катион выделяется нацело, а анион соединяется с металлом электрода и, след., образует снова соль, первоначально находившуюся в растворе. Очевидно, что количество соли, вновь образовавшейся у анода, будет эквивалентно прошедшему количеству электричества, т. е. то же количество m. Вместо уменьшения концентрация на m—а у анода произойдет увеличение концентрации на а (из m нужно вычесть m—а). Очевидно, для определения относительных чисел переносов ионов п = а/m и 1—n = (m—a)/m необходимо в этих опытах отдельно определить т. Для этой цели в цепь включают серебряный вольтметр и им определяют количество прошедшего электричества; тогда легко по закону Фарадея вычислить т. Гитторф в своих исследованиях 1853—1859 года различными методами определяет величины п и 1—n. Эти исследования легли краеугольным камнем современной электрохимии; в свое же время, отрицательное отношение самых выдающихся ученых (Магнуса, Г. Виддемана и др.), заставило автора написать следующие слова. "В моих электролитических сообщениях я взял на себя смелость критиковать и отчасти опровергать теории исследователей, высокие заслуги которых я признаю не менее их горячих приверженцев. Я никогда бы себе не позволил этой оппозиции и мои личные воззрения подчинил бы им, если бы не голые факты... Сомнение в верности этих фактов (открытый Гитторфом перенос ионов) я пытаюсь устранить тем соображением, что даже малоопытный исследователь мог бы скоро и легко доказать неверность их, и тем, что это доказательство повредило бы только мне, а не науке. Иначе обстоит дело с теориями ваших авторитетов. Как они благодетельны, если только основательны, так точно целые века разрушительно задерживают прогресс знания, если они не верны". Методы, предложенные Гитторфом для исследования относительной скорости переноса ионов весьма разнообразны. В настоящее время часто в лабораторной практике применяется метод, предложенный Нернстом и Лёбом и усовершенствованный мной. Этот метод основан на осторожном последовательном собирании после электролиза через каучуковый запор о (фиг. 3) сначала нижнего анодного тяжелого слоя — z, потом среднего — у, который должен остаться неизмененным электролизом, и в конце верхнего, катодного — x, который становится удельно легким благодаря уменьшению концентрации соли.
Фиг. 3. Прибор Кистяковского для определения переноса ионов.
Обстановка опыта видна из фиг. 3, изображающей сосуд, в котором происходит электролиз хотя бы азотносеребряной соли между серебряными электродами. Углубление r предохраняет раствор от перемешивания его отпадающими от катода кусочками серебра, выделяющегося в виде дендритов. Гарантией удачи опыта является неизменяемость среднего слоя — у.
Электропроводность растворов. Уже Гитторф высказал, что изучение электропроводности может пролить свет на явления, совершающиеся в растворе при прохождении тока. Вскоре это предположение осуществилось, благодаря систематическим исследованиям Кольраушем электропроводности и открытию закона Кольрауша - Гитторфа. С современной точки зрения, электролиты проводят ток только одновременно с молекулярными перемещениями свободных ионов. "Молекулярными" написано для того, чтобы не представлять перенос ионов, как проскок от одного электрода к другому. Явление совершается подобно диффузии одного газа в атмосферу другого газа, т. е. небольшими отрезками пути, но зато одновременно всеми ионами. Для сравнения электропроводности различных растворителей принято определять удельную электропроводность, т. е. вычисляют электропроводность кубического сантиметра любого раствора, помещенного в сосуд кубической формы, причем электродами служат противоположные стенки кубика, равные, очевидно, квадратному сантиметру. Электропроводность выражают в обратных омах, т. е. за единицу электропроводности принимают тот же ом (столб ртути длины 1,063 метра, поперечное сечение 1 кв. мм). Чтобы от сосуда произвольной формы перейти к удельной электропроводности, пользуются законом, что электропроводность сосуда с прямыми параллельными стенками уменьшается пропорционально расстоянию между электродами l и увеличивается пропорционально поперечному сечению сосуда f , если только с этим сечением совпадают по величине, форме и положению электроды. Очевидно, χ = W—1 l/f (см. обознач.). Чтобы измерить электропроводность предложено несколько методов: Бути, Кирхгофа, Кольрауша и др. Задача, которую должно преследовать при измерении электропроводности электролита, заключается в сохранении неизмененного состояния раствора у электродов при прохождении тока. Это достигнуто Кирхгофом и Кольраушем применением переменных токов. Метод для сравнения электропроводностей основан на мостике Уитстона (см. Гальванопроводность и электропроводность металлов). Успех метода Кольрауша должно приписать тому обстоятельству, что для определения отсутствия тока в соединительной ветви cd мостика Уитстона им был использован телефон. Когда в телефоне наблюдается минимум силы звука, тогда отношение сопротивлений R к W равно отношению аd к db, т. е W—1 = R ad/db; отсюда χ = R (ad/db)(l/f). Значение R отсчитывается в магазине сопротивления непосредственно числом введенных омов, отношение ad к db получается отсчетом на мостике. На фиг. 4 приведен случай, когда оно равно 42:58.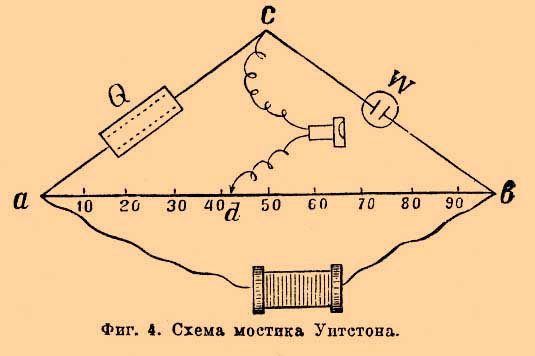
Фиг. 4. Схема мостика Уитстона.
Остается определить l/f для данного сосуда. Эти определения делаются для сосудов формы, приведенной на фигуре 5 A, В и D, пользуясь величинами удельной электропроводности растворов, определенными Кольраушем в сосудах С фигуры 5.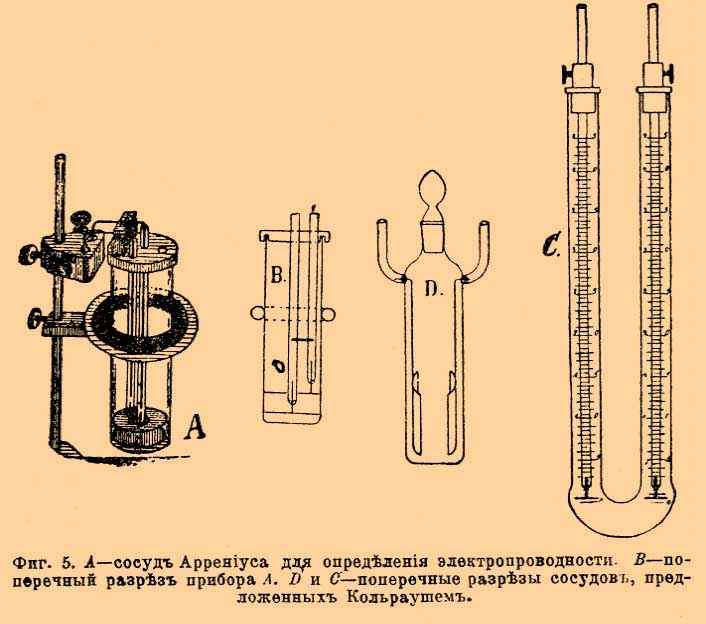
Фиг. 5. A — сосуд Аррениуса для определения электропроводности; B — поперечный разрез прибора А; C и D — поперечные разрезы сосудов, предложенных Кольраушем.
Перемещая электрод в сосуде С (фиг. 5) на расстояние l, зная начальную электропроводность и полученную после перемещения, а также поперечный разрез сосуда f, Кольрауш из уменьшения электропроводности, соответствовавшей введению столба жидкости длиной в l и поперечного разреза f, вычислял удельную электропроводность. Этим приемом Кольрауш обошел трудность полного совпадения электрода с поперечным разрезом сосуда. Определенные им точные величины для электропроводности целого ряда жидкостей послужили основанием для определения l/f, так называемой "емкости" любого сосуда. Зная χ для данной жидкости, наливая ее в любой сосуд (см. фиг. 5) и определяя R и ad/db на мостике Уитстона, вычисляют l/f для нового сосуда из уравнения χ = R(ad/db)(l/f). Фигура 6 дает общую обстановку опыта определения электропроводности. Ниже приведенная таблица дает электропроводности некоторых водных растворов в обратных омах, служащие для определения емкости сосудов.
| χ при 18° | χ при 25° | |
| Насыщенный раствор поваренной соли (NaCl) | 0,21605 | 0,25130 |
| Нормальный раствор хлористого калия (КСl) | ||
| 74,6 грамма в литре раствора | 0,09822 | 0,11180 |
| 1/10 норм. раствор КСl, | ||
| т. е. 7,46 г в литре | 0,01119 | 0,01288 |
| 1/50 нормальный раствор КСl 1,492 г в литре | 0,002397 | 0,002765 |
| 1/100 нормальный раствор КСl 0,746 г в литре | 0,001225 | |
| 0,001413 | ||
| "БРОКГАУЗ И ЕФРОН" >> "Э" >> "ЭЛ" >> "ЭЛЕ" >> "ЭЛЕК" |
Статья про "Электрохимия" в словаре Брокгауза и Ефрона была прочитана 3851 раз
| Бургер двойного помола |
| Каша со столетними яйцами |
TOP 15
- Волос
- Проно
- Степные животные
- Гимнастика
- Индийский океан
- Архитектура
- Сравнение, в литературе
- Манда
- Клитры
- Колесование
- Испарение
- Травоядные животные
- Оплодотворение у pacтений
- Вредные насекомые
- Электризация тел