
БНБ
"БРОКГАУЗ И ЕФРОН" (121188)
- Photogallery
- Естественные науки - Математика - Технология
- Авиация и машиностроение
- Высокие технологии
- Вычислительная техника
- Нанотехнология
- Роботехника
- Энергетика
- Электроника
Сталь, химический анализ
Определение "Сталь, химический анализ" в словаре Брокгауза и Ефрона
Сталь, химический анализ
Сталь, химический анализ*
— Способы химического анализа для С., чугуна и железа почти совершенно одинаковы; поэтому здесь укажем приемы анализа вообще различных сортов железа, а не специально одной С.
Анализы железа принадлежат к наиболее трудным, вследствие большего количества элементов, входящих в состав различных сортов железа, и необходимости большой точности анализов, так как многие элементы, как, например, углерод, сера, фосфор и пр. при содержании даже в ничтожных количествах значительно изменяют механические свойства железа. При той важности, которую имеет химический состав для характеристики образцов железа, очень часто данные различных лиц или лабораторий настолько отличаются между собой, что разница превышает предел, допускаемый практикой. Разница в анализах для одного и того же сорта железа зависит главным образом от того, как была взята проба для анализа, и от способа анализа. Многочисленными исследованиями вполне установлен факт, что кусок железа, полученный при застывании жидкого однородного металла, подвергнутый или нет дальнейшей обработке, в различных частях вообще имеет различный состав, не говоря уже о продуктах пудлингования, которые по самому ходу их производства являются неоднородными.
Разница в составе в различных частях железа, перешедшего из жидкого состояния в твердое, вообще тем значительнее, чем дольше длился этот переход; поэтому для определения состава приготовляемого чугуна или стали лучше всего взять при выпуске несколько небольших проб жидкого металла и вылить в форму, где он мог бы быстро остыть. Для приготовления пробы для анализа полученный слиток просверливается посередине, и опилки перемешиваются. Для анализа белого чугуна, который не поддается сверлу, берутся отдельные кусочки, измельчаются в стальной закаленной ступке и порошок хорошо перемешивается. При сверлении поверхность слитка предварительно очищается наждачной бумагой или напильником; сверло должно быть чистое и сухое. Употребление напильников для получения пробы вообще избегается, потому что обыкновенно при этом они стираются и частички их примешиваются к пробе. Опилки, в которых подозревается присутствие жира, промываются бензином и сушатся. Для сверления могут быть пригодны ручные станки, например наподобие изображенного на фиг. 1а и 1 b.
Фиг. 1 a
Фиг. 1 b
Наибольшее затруднение представляет получение средней пробы для анализа уже готового твердого металла; тогда приходится сверлить взятый образец в разных местах и в разных направлениях и смешиванием стружек получать средний образец, хотя подобный прием и малонадежен. При анализе серого чугуна советуют получать лишь столько стружек, сколько нужно для анализа, так как вместе со стружкой освобождаются частички графита, который легко может быть отсеян; поэтому для анализа берется, по возможности, вся стружка до последних следов. Благодаря трудности вполне рационально составить среднюю пробу данного образца, некоторые предлагают определять изменение состава в разных пунктах его и затем уже при помощи вычисления определять средний состав. Во всяком случае, приготовление пробы для анализа принадлежит к важнейшим операциям, от которых зависит результат анализа, и эта операция должна находиться в опытных руках, хотя, к сожалению, правила к составлению пробы намечены только в очень грубых чертах. Другая причина, от которой получаются разницы в анализе, лежит в выборе различных методов для производства одного и того же определения. В последнее время различные способы подвергались тщательному сравнению многими исследователями для выяснения причин расхождения результатов. На промышленных конгрессах по изучению железа в Германии и Англии все настоятельнее высказывается желание установить для каждого сорта железа ряд наиболее целесообразных методов анализа для различных нужд практики, т. е. делается ли анализ для контроля производства, для решения ли обычных вопросов в спорных случаях или с научной целью. Установленные методы анализа должны отличаться простотой и соответственной требованию точностью. Для наиболее важнейших определений обществом германских химиков на железоделательных заводах установлены некоторые способы, как норма. В применении этих способов, во избежание уклонений от нормального хода анализа, необходимо строго придерживаться всех мелочных указаний относительно навесок, концентраций, температур, времени нагревания и пр. Подобный рецептурный характер имеют и все вообще указания при производстве технических анализов для того, чтобы придать им необходимую для практики простоту, ясность и определенность. Укажем способы определения главнейших составных частей железа. Первое место между ними занимает углерод.
Углерод. Определение всего количества углерода. Общее содержание углерода узнается обыкновенно, сжигая его и определяя вес полученной углекислоты. Для сокращения вычислений очень часто навеску берут равной 27,27 г и тогда каждый 0,01 г полученной СО 2 отвечает 0,1% углерода в исследуемом образчике. Иногда СО 2 определяется по объему. Все способы определения общего содержания углерода можно разделить на две группы: I) сжигание углерода производится непосредственно во взятом образце без предварительного отделения, II) углерод выделяется предварительно в свободном состоянии и уже в таком виде сжигается.
I. Лучший способ по точности результатов и по скорости работы состоит в сжигании исследуемого образца мокрым путем — хромовой смесью; образовавшаяся при горении углерода углекислота поглощается едким кали или натристой известью и взвешивается. Определения удобнее всего производятся в условиях, выработанных Корлейсом и принятых на многих немецких заводах, например у Круппа в Эссене. Прибор, служащий для определения (фиг. 2), состоит из колбы A, где производится окисление образчика хромовой смесью, поглотительных приборов B и приборов C для очищения от углекислоты воздуха, который во время опыта пропускается через аппарат.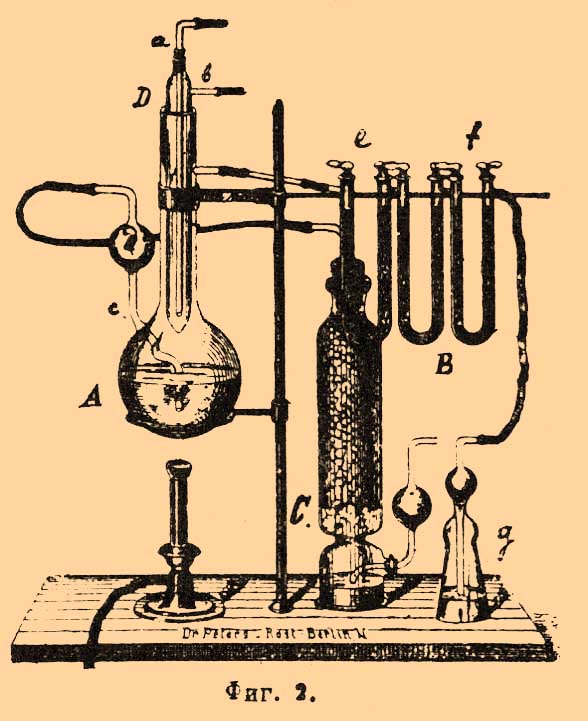
Фиг. 2.
Колба A имеет длинное горло, в которое вставляется трубка D, хорошо пришлифованная к горлу и запаянная в нижнем конце. Эта трубка играет роль пробки и вместе с тем холодильника: в трубку D вставляется тонкая загнутая вверху трубочка a, через нее пропускается ток воды, которая уходит через боковой отросток в трубке b. Хотя трубка хорошо пришлифовывается к горлу колбы, но для большей герметичности края горла несколько возвышаются над шлифом и делаются раструбом; получается таким образом небольшая воронка, в которую во время опыта наливают воды. В колбу для в пускания в нее воздуха сбоку впаяна тонкая, слегка изогнутая трубочка c, которая идет почти до дна колбы; наверху она имеет маленький шарик d, который служит предохранителем, чтобы, в случае увеличения давления газов в приборе, хромовая жидкость не могла быть тотчас переброшена в соседний прибор. Во избежание закупорки трубочка не должна быть очень тонка (не менее 6 мм). Поглотительный аппарат состоит из 3-х U-образных трубок с кранами, соединенных между собой каучуковыми трубками. Трубка e наполнена стеклообразной фосфорной кислотой и служит для поглощения паров воды, уносимых током газов из колбы, остальные же трубки содержат натристую известь и некоторое количество фосфорной кислоты (у выхода газа) для удержания воды, выделяющейся из натристой извести при соединении с углекислотой. Трубка f соединяется со склянкой g, содержащей крепкую серную кислоту; она предохраняет доступ влажности в поглотительные приборы. Для освобождения воздуха, поступающего в прибор от углекислоты, служит столбик C с натристой известью, на дне у него налит раствор едкого кали. Для производства анализа требуется: 1) насыщенный раствор хромовой кислоты (берется не химически чистая продажная хромовая кислота, содержащая обыкновенно много органических веществ, а так называемая очищенная кислота); 2) раствор медного купороса — 200 г чистой соли на 1 литр воды; 3) чистая крепкая серная кислота. Так как все эти жидкости сами по себе могут содержать следы органических веществ, то предварительно их удаляют. Опыт ведется таким образом. В колбу A вливают 25 куб. стм хромовой кислоты, 150 куб. стм раствора медного купороса и 200 куб. стм серной кислоты, взбалтывают, нагревают до кипения и кипятят около 10 минут (холодильник при этом пускается в ход). После этого горелку удаляют и в течение 10 минут пропускают через колбу ток воздуха; колба соединяется с поглотительным аппаратом, и ток воздуха поддерживается еще 5 минут. Трубки с натристой известью отделяются от прибора, взвешиваются после охлаждения и вновь ставятся на свое место. Берется навеска исследуемого образчика — от 0,5 до 5 г, смотря по содержанию в нем углерода, и в небольшой скляночке при помощи платиновой проволоки опускается в колбу. Жидкость вновь начинают кипятить, пропуская слабый ток воздуха, пока образчик не растворится вполне, на что требуется 1—2 часа. Когда операция кончилась, горелку удаляют, некоторое время пропускают через прибор воздух (около 2 литров) и взвешивают трубки с натристой известью. Описанный способ определения углерода пригоден почти во всех случаях; исключение составляют некоторые образцы, содержащие большое количество хрома или кремния, которые не вполне поддаются действию хромовой смеси. Для определения углерода может быть применен и обычный прием элементарного органического анализа — сожжение в трубке с окисью меди в струе кислорода; но он требует соблюдения многих предосторожностей, без чего результаты анализа сильно колеблются, в чугуне, например, разница может доходить до 0,643% (Ледебур). Этот последний прием определения углерода в настоящее время применяется довольно редко.
II. Для выделения углерода в свободном состоянии чаще всего употребляют медные соли (медный купорос, двойная соль хлорной меди с хлористым аммонием и пр.), которые растворяют железо, оставляя нетронутым (весь или почти весь) углерод (способы Pearce, Mc. Creath, Ulgren, Langley и пр.); в других случаях образчик накаливается в струе хлора (Велер) или в струе хлористого водорода (С. К. Девилль), причем улетучивается хлористое железо; иногда для растворения железа применяют бром и йод (Eggerz), расплавленное хлористое серебро (Берцелиус), слабую соляную кислоту в присутствии электрического тока (Binks, Weyl) и пр. Выделенный по этим способам углерод сжигается или в струе кислорода с окисью меди, или при помощи хромовой смеси. Изучение различных способов определения углерода, в основе которых лежит предварительное выделение углерода, показало, что в большинстве случаев они дают % содержание углерода ниже требуемого. Чаще всего это происходит оттого, что при выделении углерода некоторое количество его образует газообразные углеводороды, которые теряются. Потери углерода варьируют не только в зависимости от способа анализа, но и при употреблении одного и того же способа от % содержания углерода в исследуемом образчике. Приведем описание некоторых наиболее употребительных способов этого рода. Способ Пирса (Pearse) и Мак. Крита (Mc. Creath). Для приготовления двойной соли хлорной меди и хлористого аммония растворяют в воде на 107 частей нашатыря 170,3 частей кристаллической хлорной меди CuCl 22H2 O и раствор подвергают кристаллизации; полученную двойную соль растворяют (300 г ее в 1 литре воды), если нужно — фильтруют через прокаленный асбест и сохраняют в склянках со стеклянными пробками. Так как продажный нашатырь очень часто содержит примесь органических веществ, то он должен быть тщательно очищен повторной кристаллизацией. Для анализа берут навеску 1 г для чугуна и 3 г для стали, кладут ее в стакан или коническую колбу и приливают 100—200 куб. стм раствора медной соли. Жидкость энергично взбалтывают несколько минут сначала при обыкновенной температуре, а затем при нагревании не выше 60°. При большем количестве проб их ставят на водяной бане в один ряд и размешивают механически. При растворении железа сначала выделяется медь, а затем она вновь растворяется; остается углерод, сернистое, кремнистое железо и пр.; операция требует около ½ часа времени. Если выделилась основная железная соль, к жидкости прибавляют несколько капель соляной кислоты; фильтруют через прокаленный асбестовый фильтр, осадок промывают сначала раствором медной соли, а затем кипящей водой до исчезновения реакции на хлор. Остаток сжигают в печке для органического анализа, причем в трубку за слоем окиси меди помещают некоторое количество хромовокислого свинца и серебряную спиральную пластинку для удержания серы и хлора. В среднем результаты анализа по этому способу довольно удовлетворительны, хотя (по Ледебуру) разницы отдельных наблюдений для чугуна (около 4% С) доходят до 0,24%, для С. (0,4% С) до 0,04%. Выделившийся углерод сжигают иногда с хромовой смесью, для чего может служить прибор на фиг. 2; но обыкновенно здесь применяются приборы гораздо проще. Способ Ульгрена. Для растворения железа употребляется крепкий раствор медного купороса, который готовится таким образом: 200 г медной соли растворяется в воде, к раствору прибавляется по каплям слабый раствор едкого натра, пока не образуется осадок, не исчезающий при взбалтывании. Дав осадку отстояться, жидкость фильтруют через асбестовый прокаленный фильтр и разбавляют до литра. Берут навеску 1 или 3 г, кладут в стакан, прибавляют медного раствора (на каждый 1 г по 50 куб. стм) и размешивают при легком нагревании; после растворения железа остаток промывают и сжигают, как описано выше. Объемное определение. Для определения углекислоты по объему имеются способы Виборга, Фогеля, Лунге и Маршлевского, Гемпеля, Жирара и др., сущность которых заключается в том, чтобы собрать всю выделившуюся при анализе углекислоту; так как она всегда получается в смеси с воздухом, то, определив объем газа, поглощают углекислоту щелочью и, измерив вторично остающийся газ, по разности находят объем, а затем и вес углекислоты. Довольно удобен для этой цели прибор Лунге (фиг. 3).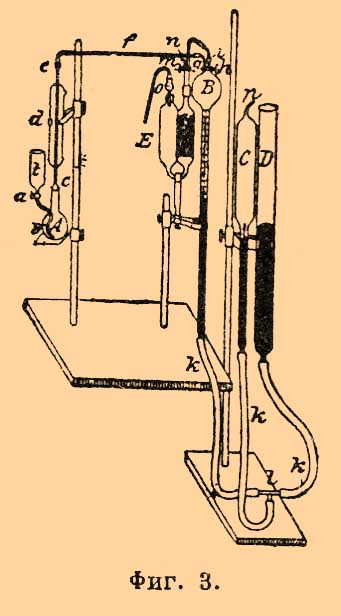
Фиг. 3.
Он состоит из трех частей: A назначена для окисления углерода, B, C, D — для измерения газа и E — для поглощения углекислоты. Колба А около 200 куб. стм снабжена воронкой t с краном a и трубочкой b, идущей до дна колбы; к ней на шлифе c пристроен холодильник d. К холодильнику сверху присоединена на шлифе e капиллярная трубочка f, ведущая к газоизмерительному прибору B; последний состоит из шара емкостью в 100 куб. стм с трубкой, разделенной на 1/10 куб. стм; на шаре двухходовой кран h. При помощи толстостенных каучуков k и стеклянного тройника l к измерительному прибору B присоединены открытая трубка D и цилиндрический запаянный сверху сосуд C, емкостью 100 куб. стм и с такой же градуированной трубкой, как при B. Все эти части наполнены ртутью и зажаты в штативах. В сосуде C находится насыщенный водяными парами воздух; количество его такое, что в сухом состоянии при 0° и 760 мм давления он занимает 100 куб. стм. При помощи этого приспособления очень легко приводить объем газа, находящегося в B, к 0° и 760 мм (газ предполагается насыщенным водяными парами), не пользуясь барометром и термометром и не делая сложных вычислений (см. Нитрометр). Шар B при помощи каучуковых смычек g и i соединяется с трубочкой f и пипеткой E; последняя такая же как в приборе Орса для газового анализа (см). Она наполнена крепким раствором едкого кали и стеклянными трубочками (в одной половине для увеличения поверхности соприкосновения щелочи с газом); у пипетки двухходовой кран, дающий возможность сообщать ее с внешней атмосферой. O — трубочка с натристой известью. При анализе, по Лунге, навеска кладется в колбу A, обливается медным купоросом; растворение железа производится при обыкновенной температуре при взбалтывании. Когда растворение кончилось (через 1—6 часов), в колбу вставляют холодильник d и, соединив ее с шаром B, выкачивают из нее воздух, поднимая и опуская трубку D (до 6 раз). Через воронку t приливают затем в колбу A хромовой смеси, нагревают сначала осторожно, а под конец кипятят около ½ часа. Газ собирается в B. Удалив горелку, вливают в колбу через воронку t 1— 2 куб. стм перекиси водорода; образовавшийся кислород вытесняет растворенную в жидкости углекислоту; затем вливают в колбу горячей воды до крана h и вытесняют весь газ в B. Если его мало, всасывают в B через кран m и n немного воздуха, лишенного углекислоты. Измерив объем газа, переводят его в пипетку E, а из нее вновь в шар B для измерения. Каждый 1 куб. стм углекислоты при 0° и 760 мм отвечает 0,536 углерода. Количество раствора медного купороса, хромовой и серной кислот, которые берутся Лунге, варьируют, в зависимости от % содержания углерода, по следующей таблице:
| Углерод, % | Навеска | Медный купорос (куб. стм) | Хромовая кислота, 100 г в 100 ч. воды (куб. стм) | Серная кислота (куб. стм) | Перекись водорода | ||
| Удельный вес 1,65 | Удельный вес 1,71 | Удельный вес 1,10 | |||||
| выше 1,5° 1,5—0,8° 0,8—0,5° 0,5—0,25° ниже 0,25° | 0,5 1 2 3 5 | 5 10 20 50 50 | 5 10 20 45 50 | 135 130 130 | — — — 75 70 | 30 25 5 5 5 | 1 2 2 2 2 |
Объем газа при первом измерении должен быть для чугуна не менее140 куб. стм, а железа не менее 130 куб. стм, иначе, после извлечения углекислоты и при вторичном измерении газа, уровень ртути в измерительном приборе установится в самом шаре, а не в градуированной трубке.
Способ Эггерца. Для определения химически связанного углерода (карбидного) и углерода закалки, главным образом в С., Эггерц предложил в 1862 г. очень простой колориметрический способ, который быстро вошел во всеобщее употребление. Сущность его заключается в том, что химически связанный углерод при растворении различных образчиков железа в крепкой азотной кислоте дает темно-бурые растворы, густота окраски которых (при одинаковых объемах полученных растворов) пропорциональна содержанию углерода в исследуемых образцах; если же соответственным разбавлением получать растворы одинаковой окраски, то содержание углерода прямо пропорционально объемам растворов. Благодаря этому обстоятельству, ход анализа прост. Берутся две одинаковых навески: одна — с определенным % содержанием углерода a, a другая с неизвестным x; после растворения образчиков в азотной кислоте и разбавления растворов до одной и той же густоты окраски, пусть объем первого раствора будет A и второго B, тогда x:a=B:A. Способ, очевидно, не может быть применен в присутствии никеля, хрома, а также и меди в таких количествах, которые могут влиять на окраску растворов. Для определения углерода по Эггерцу прежде всего выбирают эталоны, типические образцы, с которыми должны сравниваться все исследуемые сорта железа. Количество типических образцов бывает различно, смотря по характеру анализов. Одно нужно сказать, желательно, чтобы выбираемые для сравнения типические образцы были одного происхождения с исследуемыми и близко подходили к ним по химическому составу и по физическим свойствам, например бессемеровская С. должна сравниваться с бессемеровской, тигельная с тигельной, мартеновская с мартеновской и пр. Дело в том, что оттенки растворов получаются несколько разные, в зависимости от качеств химически соединенного углерода, например, углерод закалки дает более светлые растворы, чем карбидный, поэтому закаленную С. для испытания по Эггерцу отпускают и сравнивают с отпущенной С. и пр. Испытание ведется таким образом. Берут навески по 0,1 г и исследуемого образца, и соответственного типического образчика — эталона, помещают в пробирки (15 мм ширины и 120 мм длины) и обливают определенным количеством азотной кислоты удельного веса 1,2, которое зависит от % содержания искомого углерода. Для содержания ниже 0,3% берут 3 куб. стм кислоты, для 0,3%—0,5% — 3,5—4 куб. стм; для 0,5%—0,8% — 4—5 куб. стм; для 0,8%—1% — 6 куб. стм и для содержания химически соединенного углерода выше 1% — 7 куб. стм. Азотная кислота должна совершенно не содержать хлора, иначе образуется хлорное железо, которое своей окраской будет вредить точности определения. Азотная кислота приливается из бюретки постепенно, и для усмирения реакции пробирки иногда ставят в холодную воду; для этой же цели образчики не должны быть очень измельчены. При растворении железа выделяются органические вещества в виде хлопьев, для растворения коих пробирки (прикрыв их) ставят в водяную баню и нагревают при 100°, по временам встряхивая для удаления осадка со стенок. Нагревают 20—45 минут, смотря по содержанию углерода. Слишком долго не следует нагревать, так как цвет растворов начинает слабеть. Когда хлопья исчезли и прекратилось выделение газов, пробирки помещают в холодную воду, защищая от действия света (в особенности прямого солнечного), так как на свету окраска растворов слабеет. Если % содержание углерода неизвестно даже приблизительно, то для растворения берут сначала 3 куб. стм азотной кислоты; если при нагревании будет получаться очень темный раствор или количество органических веществ, выделяющихся в виде хлопьев, будет значительно, нужно еще прибавить азотной кислоты. Небольшой избыток азотной кислоты не вредит. Для сравнения оттенков растворов служат градуированные трубки емкостью в 30 куб. стм с делением на 0,05 куб. стм. Трубки должны быть одного диаметра (около 15 мм) со стенками одной и той же толщины и должны быть приготовлены из хорошего, неокрашенного стекла. Раствор взятого эталона выливается в градуированную трубку, пробирка ополаскивается небольшим количеством воды, которая присоединяется к раствору; последний затем разбавляется водой, так, чтобы объем всего раствора был не менее 8 куб. стм (при таком разбавлении не заметна окраска, производимая азотно-железной солью). Обыкновенно раствор разбавляют так, чтобы 1 куб. стм его отвечал 0,1% или 0,05% углерода. Затем подобным же образом выливают в другую градуированную трубку раствор исследуемого образца и постепенно разбавляют водой до получения одного и того же цвета. После каждой прибавки воды раствор хорошо взбалтывается. Уравнивание оттенков растворов составляет существенную часть операции. Для сравнивания трубок употребляются иногда особые камеры. Они состоят из деревянного ящика высотой около 90 мм, шириной на одном конце 38 мм, а на другом 127 мм; длина ящика около 610 мм; внутри он выкрашен в черный цвет. Ящик прикреплен к штативу и поднимается или опускается по желанию. В переднем конце в него вставлено матовое стекло и в крышке сделаны отверстия для трубок; крышка покрывается черной материей. Для однообразия результатов многие предпочитают делать подобные наблюдения с искусственным освещением в темной комнате; тогда к матовому стеклу присоединяют другое стекло, слегка окрашенное в синий цвет, для уничтожения окраски искусственного освещения. При анализе белого чугуна, по способу Эггерца, берут навеску 0,05, растворяют в 7 куб. стм азотной кислоты, разбавляют до 20 куб. стм и сравнивают оттенки, по возможности, быстрее, так как растворы скоро мутнеют, выделяя органические вещества. При анализе серых чугунов, а иногда и С. — выделяется графит, который изменяет оттенок раствора; для освобождения от него раствор фильтруют через асбестовый фильтр. При анализе по Эггерцу, Бриттон предложил иметь для сравнения постоянные растворы — эталоны; но они оказались неудобными для хранения. По Эггерцу, лучше всего их готовить по рецепту профессора Экмана: из хлорного железа, хлорной меди, хлористого кобальта и соляной кислоты. Получают серию растворов, отвечающих по окраске известному содержанию углерода в 1 куб. стм от 0,1% до желаемого, варьируя, например, через 0,002%. Каждого раствора наливают по 10 куб стм в градуированную трубку и для верности сравнивают их оттенки с растворами эталонов; исследуемый образец растворяется, как указано раньше, и разбавляется до 10 куб. стм; затем определяют к какому из эталонов он подходит по цвету; допустим, что взят эталон, отвечающий 0,032% углерода в 1 куб. стм, тогда искомое содержание углерода в образчике равно 0,32%. Стид (St e ad) предложил другой способ для определения химически связанного углерода, основанный на том, что углеродистые вещества, образующиеся при растворении железа в азотной кислоте, могут растворяться в едких щелочах, образуя растворы, которые гораздо более интенсивно окрашены, чем у Эггерца.
Сера. Для определения серы существует три рода способов: 1) выделение в виде сероводорода, 2) непосредственное окисление в серную кислоту и 3) окисление после предварительного удаления железа.
Исследуемый образец растворяется в слабой соляной или серной кислоте; сера выделяется в виде Н 2 S, который пропускается через соответственные поглотительные растворы. В одних случаях для этой цели применяют окисляющие вещества (например, бром, марганцово-калиевую соль, перекись водорода и пр.), которые переводят сероводород в серную кислоту; последняя затем определяется обычным путем в виде сернокислого бария; в других случаях сероводород поглощается растворами солей, дающих нерастворимые сернистые соединения (азотно-серебряной, медным купоросом, уксуснокислым кадмием или цинком и пр.). Сера определяется или по весу полученных сернистых металлов, или они окисляются, и сера определяется в виде сернокислого бария и т. п. Однако, исследования последнего времени показали, что не вся сера выделяется в виде сероводорода при растворении образца: некоторая часть ее оказывается иногда в нерастворимом остатке вместе с графитом и пр.; некоторая же часть образует летучие продукты (СН 3)2 S, проходящие без изменения через поглотительные приборы. Если не принимать во внимание этих обстоятельств, могут получаться значительные ошибки (до 50%). Опыт показывает, что при пропускании (СН 3)2 S в избытке водорода через накаленную трубку, вся сера выделяется в виде сероводорода. На этом основан способ определения серы Ролле-Кампредона. Прибор их состоит из конической колбы в 500 куб. см ёмкости, в которой происходит растворение железа. Эта колба с одной стороны соединена с Кипповским аппаратом для приготовления водорода и углекислоты, а с другой — с фарфоровой трубкой, накаливаемой в печке и с поглотительными склянками. В горло конической колбы вставлена пробка с тремя дырами; в одну входит воронка для приливания кислоты, в другую — холодильник и в третью — газопроводная трубка. Для очищения водорода от сернистого и мышьяковистого водорода служат три склянки. Первая из них содержит 150 куб. стм 2% раствора азотно-серебряной соли, вторая — 100 куб. стм того же раствора и последняя 50 куб. стм дистиллированной воды для удержания следов азотно-серебряной соли, уносимых током газов. Фарфоровая трубка имеет 600 мм длины и 33 мм в диаметре, при толщине стенок в 3 мм; трубка глазурована вся внутри, а снаружи только на концах, которые оттянуты, чтобы удобнее соединять ее каучуковыми трубками с другими частями прибора; ее защищают от резких изменений температуры, окружая асбестом. В одной поглотительной склянке находится 200 куб. стм уксуснокислого цинка для поглощения сероводорода; о полноте поглощения его судят по другой склянке, в которую наливают 50 куб. стм того же раствора (25 г соли на 1 литр воды, подкисленной 1 куб. стм уксусной кислоты). Все каучуковые трубки, служащие для соединения различных частей прибора, и каучуковые пробки должны быть из невулканизованного каучука или же выварены в 10% едком натре. Опыт ведется таким образом. Берут навеску в 5 грамм, кладут ее в колбу и вытесняют воздух из прибора током углекислоты; затем, прекратив ток углекислоты, приливают 60 куб. стм разбавленной соляной кислоты (1:2 по объему) и 30 куб. стм разбавленной серной кислоты (1:4 по объему) через воронку в колбу. Растворение несколько минут идет на холоде, а затем колбу начинают подогревать, пропуская водород; к этому времени фарфоровая трубка должна уже быть накалена докрасна. Для растворения требуется 10—30 мин.; когда оно кончилось, прибор продувают током углекислоты, во избежание взрыва при последующих определениях. Жидкости из поглотительных склянок сливают вместе и образовавшийся сернистый цинк определяют титрованием. Для этой цели к жидкости приливают некоторый избыток титрованного раствора йода. Йод выделяет серу из сернистого цинка: ZnS+I 2=ZnI2 +S. 1 г йода выделяет 32/25,37 г серы. Реакция идет довольно быстро при размешивании. Избыток йода определяется титрованием гипосульфитом натрия в присутствии крахмала. Раствор йода готовят такой крепости, чтобы 1 куб. стм его отвечал 0,001 г серы; титр его определяется в присутствии серно-цинковой соли, чтобы ближе находиться к условиям опыта. Содержание серы в сернистом цинке определяется и весовым путем: к жидкости приливают 10 куб. стм уксусной кислоты, содержащей 10% брома; сернистый цинк тотчас окисляется в сернокислый; жидкость сливают в баллон грушевидной формы, кипятят для удаления брома и осаждают сернистую кислоту в виде сернокислого бария. На многих заводах в ходу старые, менее точные способы, в которых не принято мер против потерь от образования газообразных сульфидов, проходящих без изменения через поглотительные приборы. До последнего времени в большом распространении способы, основанные на окислении сероводорода бромом:
Н 2S + 4 Вr 2 + 4H2 O = Н 2 SО 4 + 8НВr.
Способ Джонстона и Ландольта. Прибор, применяемый здесь (фиг. 4), состоит из колбы A в 750 куб. стм, где ведется разложение, Кипповского аппарата B для добывания углекислоты (она очищается, проходя раствор сулемы в a) и трубки C, где происходит окисление сероводорода. Эта трубка длиной около 60 стм, шириной около 2 стм, наполнена стеклянными бусами, смоченными соляной кислотой, насыщенной бромом. Сверху в нее вставлена воронка b, содержащая бромный раствор, и отводная трубочка d, внизу находится кран g; к ней припаяна под острым углом трубочка с краном, соединяющаяся с отводной трубкой колбы.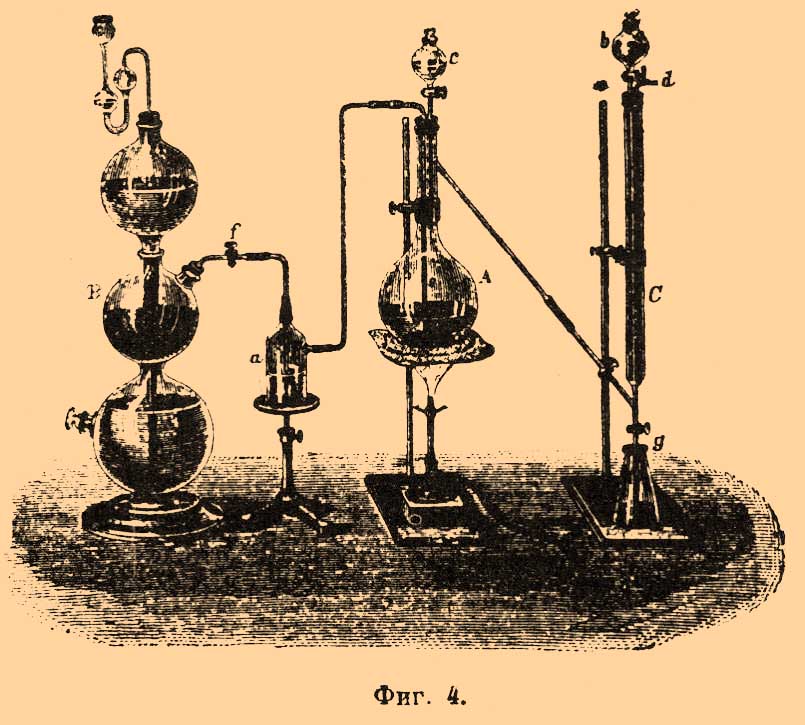
Фиг. 4.
Навеска берется 5—10 грамм и кладется в колбу, в которую наливается вода (около 100 куб. стм), так, чтобы газопроводная трубочка e погружалась в нее; смачивают трубку C бромным раствором и через воронку c приливают постепенно в колбу A 5—10 куб. стм крепкой соляной кислоты. Выделяющийся газ проходит трубку C и бромный раствор начинает обесцвечиваться. Когда это сделается заметным, открывают кран g, спускают жидкость в подставленную коническую колбу и в трубку C медленно приливают нового раствора. Когда железо растворится и газ перестанет выделяться, в колбу A приливают еще кислоты и жидкость начинают постепенно нагревать до кипения, пропуская ток углекислоты; затем нагревание прекращают, бромный раствор спускают в коническую колбу и трубку C обмывают водой несколько раз. Собранную жидкость выпаривают в фарфоровой чашке для удаления брома, разбавляют водой, фильтруют и осаждают хлористым барием обычным образом. Вместо поглотительного прибора C, иногда просто употребляют дрекселевскую склянку, в которую наливают 50 куб. стм бромного раствора. Вместо брома берется раствор марганцово-калиевой соли, аммиачный раствор перекиси водорода или перекиси натрия. Не нужно забывать, что перекись водорода почти всегда содержит серную кислоту.
Способы непосредственного определения серы состоят в том, что железо растворяется, причем сера окисляется в серную кислоту; последняя осаждается хлористым барием. Выполнение требует большой опытности, так как приходится определять серную кислоту в присутствии громадного избытка посторонней соли, а в этих условиях возможно увлечение сернокислым барием в осадок посторонних примесей. Навеска берется 5 г; для растворения одни берут крепкую азотную кислоту (40 куб. стм). Операцию ведут в стакане, который иногда приходится охлаждать при очень бурной реакции; иногда при С. с высоким содержанием углерода для начала реакции нужно к азотной кислоте прибавить немного соляной и жидкость нагреть. Некоторые смешивают навеску с бертолетовой солью и растворяют в крепкой азотной кислоте, к которой прибавлено немного брома (6 г навеска, 1 г бертолетовой соли и 50 куб. стм азотной кислоты, содержащей 1 куб. стм брома). Применяют для растворения и царскую водку и пр. Когда растворение кончилось, жидкость выпаривают досуха с соляной кислотой, прибавляют вновь соляной кислоты 20—30 куб. стм и выпаривают до тех пор, пока раствор не примет вид сиропа; его разбавляют водой и фильтруют. Раствор в холодном состоянии осаждают хлористым барием, оставляют стоять при комнатной температуре 12 часов, фильтруют и промывают холодной водой, подкисленной соляной кислотой. Как осаждение, так и промывание делается на холоде потому, что в этих условиях гораздо меньше получается железной соли в осадке. После прокаливания слегка буроватый оттенок сернокислого бария может указать на присутствие в нем следов железа; чтобы удалить его, осадок обрабатывается соляной кислотой, избыток ее выпаривается, затем к осадку прибавляют воды, нагревают до кипения и приливают несколько капель хлористого бария; тогда получается совершенно чистый сернокислый барий.
Колориметрический способ Виборга. Этот способ основан на окрашивании в желтый цвет от действия сероводорода бумажной ткани, смоченной уксуснокислым кадмием. Интенсивность окраски зависит от содержания серы в исследуемом образце; сравнивая полученную окраску с другими, отвечающими определенным количествам серы, можно вычислить % содержание серы в анализируемом образце. Аппарат Виборга (фиг. 5) состоит из круглодонной и широкогорлой колбы a, в которую вставлена пробка с двумя отверстиями; в одно входит вороночка b, а в другое широкая трубка C, с оттянутым концом. Трубка C имеет наверху закраины, на который кладется кружок из бумажной ткани, смоченный 5% раствором уксуснокислого кадмия. Чтобы герметично прижать кружок к краям трубки C, он кладется между двумя резиновыми кольцами d и сверху налегает на него деревянное кольцо e, которое прижимается к закраинам C особыми зажимами.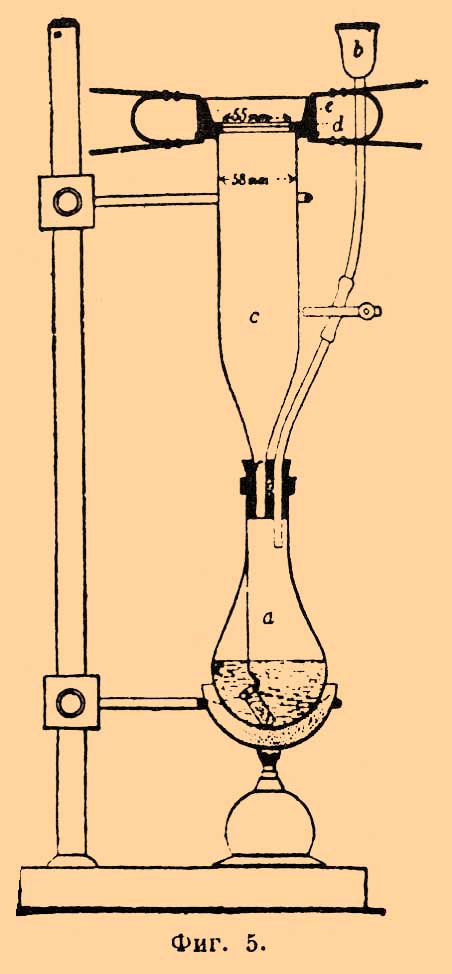
Фиг. 5.
Берут навеску 0,02—0,8 г; в колбу наливают воды и нагревают до кипения, чтобы выгнать из неё воздух. Затем или бросают в нее навеску, или опускают на платиновой проволоке в цилиндре для взвешивания. Продолжая слегка кипятить воду, вставляют пробку a, укладывают чувствительный кружок на трубке C и приливают немного (8—10 куб. стм) разбавленной серной кислоты (1:3). Когда навеска растворилась, жидкость кипятят еще несколько минут для выделения сероводорода, затем кружок снимается, высушивается и сравнивается со шкалой оттенков. Чтобы окраска кружка была равномерна, необходимо, чтобы нижнее отверстие трубки C было как раз в центре; если на кружке окажутся складки, н
| "БРОКГАУЗ И ЕФРОН" >> "С" >> "СТ" >> "СТА" >> "СТАЛ" |
Статья про "Сталь, химический анализ" в словаре Брокгауза и Ефрона была прочитана 3174 раз
| Коптим скумбрию в коробке |
| Кетчуп из бананов |
TOP 15
- Волос
- Проно
- Степные животные
- Гимнастика
- Индийский океан
- Архитектура
- Сравнение, в литературе
- Манда
- Клитры
- Колесование
- Испарение
- Травоядные животные
- Оплодотворение у pacтений
- Вредные насекомые
- Электризация тел