
БНБ
"БРОКГАУЗ И ЕФРОН" (121188)
- Photogallery
- Естественные науки - Математика - Технология
- Авиация и машиностроение
- Высокие технологии
- Вычислительная техника
- Нанотехнология
- Роботехника
- Энергетика
- Электроника
Нитрометрия
Определение "Нитрометрия" в словаре Брокгауза и Ефрона
Нитрометрия
Нитрометрия*
— Н., или азотометрией, называется совокупность способов количественного определения азота. При анализе азот определяется в виде: I) свободного газа, II) аммиака и III) в виде окиси азота или азотной кислоты. Выбор того или другого способа зависит от многих условий.
I) Определение в виде свободного азота. При анализе органических соединений, содержащих азот, когда требуется наибольшая тщательность в определении, применяется метод Дюма. Он состоит в том, что органическое вещество сжигается обычным образом с окисью меди (см. Органический анализ); могущие образоваться при этом окислы азота пропускаются над слоем накаленной меди, которая разлагает их, выделяя азот; последний тщательно собирают, вытесняя его током углекислоты и измеряют. Опыт ведется следующим образом. В стеклянную тугоплавкую трубку для органического анализа от 80 до 90 см длиной, запаянную с одного конца, кладут слой в 12 — 15 см чистого магнезита MgCO 3 или двуугленатриевой соли NaHCO 3, хорошо высушенной над H 2SO4, и поверх него асбестовую пробку. MgCO 3 и NaHCO 3 служат для выделения углекислоты. Затем следует слой в 10 см окиси меди; потом кладут взятое вещество; если оно твердо, — его быстро и хорошо смешивают с некоторым количеством порошковатой окиси меди в ступочке, которую, как и стенки трубки, если можно так выразиться, ополаскивают порошком той же окиси меди, чтобы на них не осталось следов анализируемого вещества; если вещество жидко, его берут в стеклянной ампулке. Вслед за анализируемым веществом идет слой окиси меди в 40 см, асбестовая пробка и пробка из чистой медной сетки, свернутой плотной спиралью, длиной в 15 — 20 см. Последнюю проще всего готовят таким образом, что свернутую надлежащим образом сетку из красной меди прокаливают на паяльном столе, слегка окисляя ее, чтобы сжечь все имеющиеся на ее поверхности горючие вещества, и в накаленном состоянии опускают в пробирку, в которую налито несколько капель метилового спирта. Пары спирта, которые при этом вспыхивают, и восстановляют медь. Перед употреблением для анализа сетку высушивают при 120° — 130°. Открытый конец трубки соединяют с прибором для собирания азота. Удобнее всего для этой цели прибор (фиг. 1).
Фиг. 1. Прибор для собирания азота при анализе.
Он отличается от обыкновенной газовой бюретки Гемпеля (см. Лаборатория) тем, что имеет внизу (на фиг. справа) отросток, который и соединяется с трубкой, для сожигания. На дне бюретки налит слой ртути на несколько мм выше нижнего отверстия упомянутого отростка, и вся она, так же как и шар, наполнена крепким раствором едкого кали; ртуть здесь служит запором. Опыт ведется таким образом. Соединив прибор с трубкой для сжигания, открыв кран (наверху) и опустив шар так, чтобы по возможности удалить щелочь из бюретки, слегка нагревают MgCO 3 или NaHCO 3, чтобы вытеснить углекислотой из трубки весь воздух; в этом убеждаются, заполняя бюретку по временам щелочью и наблюдая, сполна ли поглощается выделяющийся газ. Когда это будет достигнуто, умеряют выделение СО 2, бюретку вместе с капилляром окончательно наполняют щелочью; закрывают кран и приступают к сжижению. Сначала накаливают медную сетку, затем окись меди в передней и в задней части трубки, постепенно приближаясь к анализируемому веществу и заботясь, чтоб оно не начало разлагаться прежде, чем окись меди будет накалена (подробности см. Органический анализ). При определении азота во взрывчатых веществах сожжение должно вести крайне осторожно, иначе быстро образовавшийся газ пройдет несгоревшим через сожигательную трубку; поэтому для последних способ Дюма редко применяется. Когда сожжение кончилось, усиливают выделение СО 2, чтобы выгнать весь азот из трубки в бюретку. Ток углекислоты поддерживают до тех пор, пока объем газа в бюретке не перестанет увеличиваться. Полученный азот измеряют обычным образом (см. Газовый анализ), переводя его в измерительные трубки над водой. Если объем его V, атм. давл. Н, темп. t и давл. водяных паров при этой темп. = b, то вес полученного азота p=[V(H—b)0,001256]/[760(1+0,00367t)]. Необходимо поверочными опытами убедиться в чистоте полученного азота; закись азота здесь не вредит, так как объем ее равен объему находящегося в ней азота. Присутствие окиси азота узнается при помощи железного купороса.
Существует много видоизменений метода Дюма. Симпсон, чтобы облегчить сожжение, советует брать окись ртути: он прибавляет некоторое количество ее к магнезиту (2 г на 12 г магнезита) и к окиси меди, с которой смешивается взятое вещество (4 ч. HgO на 5 ч. СuО). Чтобы облегчить получение углекислоты, некоторые берут смесь соды с двухромово-калиевой солью, другие же просто ведут сожжение в открытой трубке, соединенной с каким-либо прибором для добывания углекислоты, в чистоте которой нужно предварительно убедиться.
Для определения азота в аммиаке, аммиачных солях, мочевине и т. п. иногда удобно пользоваться способностью их разлагаться бромноватистыми или хлорноватистыми щелочами, напр.
3NaBrO + 2NH3 = 3NaBr + 2N2 + 3H2 О
(см. Мочевина). Определение азота в газах — см. Газовый анализ.
II) В виде аммиака азот определяется глав. образом: а) по способу Вилля и Варрентраппа и b) по способу Кьельдаля; как тот, так и другой способ прилагаются исключительно к органическим веществам.
а) По способу Вилля и Варрентраппа исследуемое вещество прокаливается в смеси с натристой известью (см.); горение совершается на счет кислорода натристой извести; водород которой с азотом взятого вещества дает аммиак; последний поглощается серной или соляной кислотой и определяется титрованием или в виде хлороплатината. Опыт ведется таким образом. Берется тугоплавкая стеклянная трубка, как для органического анализа, но лишь около 50 см длиной; задний конец ее оттянут, загнут кверху и запаян. В нее насыпают слой около 5 см зерненой натристой извести, которую перед опытом слегка прокаливают, чтобы удалить часто бывающую в ней примесь аммиачных солей. Вложив асбестовую пробку, насыпают далее в трубку смесь взятого вещества (0,2 — 0,4 г) с порошковатой натристой известью, которой берут столько, чтобы получился в трубке слой в 15 см. Как ступочку, в которой производилось смешение вещества с натристой известью, так и стенки трубки ополаскивают порошком натристой извести и, дополнив трубку зерненой натристой известью, вставляют асбестовую пробку. Слой натристой извести не доходит примерно см 5 до конца трубки. Трубку соединяют с прибором для поглощения аммиака, состоящим или из обыкновенной V-образной трубки, или из трубки Вилля и Варрентраппа (фиг. 2).
Фиг. 2. Прибор для поглощения аммиака.
Здесь находится известный объем титрованной серной кислоты (если определяют аммиак титрованием) или слабая соляная кислота (уд. вес около 1,1). Кислота наполняет только нижнюю часть трубки, чтобы ее не выбросило током газа или не всосало в трубку для сожжения при сильном поглощении аммиака. Трубку для сожжения начинают накаливать обычным образом спереди, постепенно приближаясь к взятому веществу и постоянно регулируя быстроту разложения его. Избегают сильного накаливания, чтобы не разложить NH 3. Ток газов через прибор для поглощения не должен быть очень быстрый, иначе аммиак не поглотится сполна. Когда вещество сгорело окончательно и ток газов прекратился, отламывают у трубки для сожжения оттянутый кончик и пропускают через нее воздух, чтобы выгнать последние следы аммиака. Если определение аммиака производится титрованием, то взятую для поглощения кислоту титруют обычным путем и определяют, сколько ее пошло на насыщение аммиака. Для получения хлороплатината, жидкость сильно концентрируют, прибавляют к ней хлорной платины, промывают спиртом, эфиром и прокаливают. По весу полученной платины вычисляют азот, принимая, что 194,8 г платины = 28 г азота. Способ Вилля и Варрентраппа имеет довольно ограниченное применение. Одни вещества дают в этих условиях вместе с аммиаком и летучие амины, другие же не разлагаются сполна; он не годится для определения азота в азо-, диазо-, нитро- и нитрозосоединениях, в некоторых белковых телах и пр. К веществам многоазотным рекомендуется прибавлять сахар или безводную щавелевую кислоту, чтобы избежать неудобств, сопряженных с выделением большого количества аммиака разом.
b) Способ Кьельдаля не требует ни специального измельчения вещества, ни печей для сожжения и дает возможность одновременно вести несколько определений. В особенности он удобен для жидких веществ. Органическое вещество разлагается здесь нагреванием с крепкой серной кислотой, а реакция доводится до конца прибавлением марганцево-калиевой соли. При этом азот взятого вещества дает серно-аммиачную соль, откуда щелочью выделяют аммиак, который определяется подобно тому, как описано выше. Опыт ведется таким образом. Берут баллон из тугоплавкого стекла емкостью около 100 куб. см с длинным горлом, в него помещают взятое вещество (от 0,3 г до 1 г, смотря по богатству азота), приливают 10 куб. см крепкой серной кислоты, не заключающей аммиачных солей, и постепенно нагревают его на голом огне (на сетке) почти до кипения, придав баллону наклонное положение, чтобы не было потерь от разбрызгивания. Кислота сначала чернеет, потом мало-помалу обесцвечивается. Тогда нагревание прекращают и в колбу осторожно бросают понемногу марганцево-калиевой соли, пока жидкость не станет окрашиваться в зеленый цвет. Осторожно нагрев баллон еще раз в течение 5 мин., ему дают окончательно охладиться; содержимое его вливают в другой баллон емкостью в 600 — 700 куб. см, прибавляют избыток раствора едкого натра (уд. в. 1,3), несколько кусочков цинка и, соединив с прибором для поглощения аммиака, кипятят около 1/2 часа. Цинк назначается, главным образом, для облегчения кипения. При кипячении принимают все предосторожности относительно того, чтобы вместе с аммиаком не попал в поглотительные приборы едкий натр (если аммиак определяется титрованием, как это обыкновенно делается). Для этого баллон ставится наклонно, отводная трубка его сначала поднимается вверх, а затем загибается книзу; для удержания брызг кипящей жидкости трубка, кроме того, снабжена вверху шаром. Одни употребляют холодильники, другие нет (напр. фиг. 3).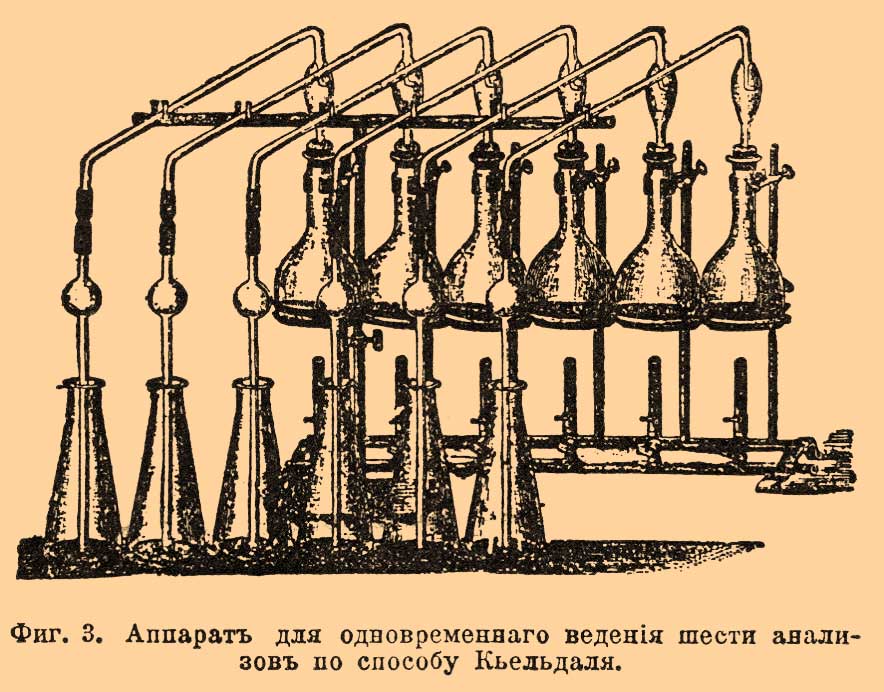
Фиг. 3. Аппарат для одновременного ведения шести анализов по способу Кьельдаля.
Кислота, служащая для поглощения аммиака, наливается в эрленмейеровскую коническую колбу, и в нее погружается кончик отводной трубки. При этом применяют обычные предосторожности, чтобы кислота не была всосана в баллон при быстром поглощении. Необходимо предварительным опытом убедиться, что взятая для разложения органического вещества кислота не содержит аммиачных солей. Для этой цели берут пробу около 10 куб. см и выделяют из нее NH 3 вышеописанным образом и, если аммиак там находится, то делают при анализе соответственную поправку. Способ Кьельдаля, подобно способу Вилля и Варрентраппа, имеет ограниченное применение, хотя исключения при нем представляются гораздо реже, Он, по-видимому, не применим для азо-, диазо-, нитро-, нитрозосоединений, азотно-кислых солей, и вообще в каждом частном случай необходимо убедиться в его пригодности. В способ Кьельдаля введено много видоизменений. Для ускорения реакции разложения берут серную кислоту, содержащую фосфорный ангидрид (до 20 — 25%). Вильфарт нашел, что разложение идет легче в присутствии окиси ртути и меди, и советует прибавлять их; то же самое достигается, по Арнольди, если на 1 г вещества брать 0,5 г безводного медного купороса, 1 г ртути и 20 куб. см серной кислоты с фосфорным ангидридом. Перед прибавкой щелочи ртуть удаляется из раствора прибавлением некоторого количества сернистого натрия. Сделано много попыток расширить сферу применения способа Кьельдаля, сделать его пригодными для определения азота в нитросоединениях, азотно-кислых солях и пр. Очень недурные результаты получаются по способу Цодльбауера. Около 0,5 г вещества, очень хорошо измельченного, он обрабатывает 30 куб. см серной кислоты, содержащей в растворе 1,2 г карболовой кислоты и 0,4 г фосфорного ангидрида, взбалтывает при охлаждении пока все не растворится, затем прибавляет постепенно 3 — 4 г цинка в порошке и оставляет на холоде около 2 часов; после этого прибавляет 0,7 г ртути и продолжает по Кьельдалю. Предполагается, что если присутствуют азотно-кислые соли или эфиры, то они разлагаются серной кислотой, выделившаяся при этом азотная кислота образует нитрофенол, который восстановляется цинком в амидофенол, последний же далее разлагается до аммиака. Если при этом присутствуют нитросоединения, то многие из них тоже восстановляются в этих условиях, и результаты анализа получаются удовлетворительные. При неудаче можно вести восстановление нитрогруппы йодисто-водородной кислотой в присутствии фосфора. Около 2 г фосфора растворяют в 20 — 25 куб. см сернистого углерода в колбе около 250 куб. см емкостью, кладут 12 г йода и выпаривают на водяной бане. Кладут сюда же навеску вещества и приливают около 8 куб. см воды. Когда реакция произошла, приливают на холоде серной кислоты и ртути и поступают, как обыкновенно (избыток йода и йодистый фосфоний, образующийся при реакции, удаляются нагреванием).
III) Определение азота в виде азотной кислоты или ее солей — см. Йодометрия и Крепкая водка; для других определений пользуются главным образом способами Шлезинга и Лунге; как тот, так и другой служат для определения азота в азотно- и азотисто-кислых солях, эфирах и тому подобных соединениях. Способ Шлёзинга основан на раскислении азотной кислоты и высших окислов азота до окиси азота NО посредством закисных солей железа; он претерпел много видоизменений. Для анализа селитры удобен следующий прием. Берут колбу емкостью 250 — 300 куб. см, заткнутую пробкой с 2 отверстиями: в одно входит шарообразная воронка с краном (нижний конец ее доходит до средины колбы и оттянут), в другое — газоотводная трубка; свободный конец последней загнут кверху и погружен в ванну с водой. В колбу приливают примерно 10 куб. см раствора хлористого железа (400 FeCl 2 на 1 литр воды) и столько же 10%-й соляной кислоты и кипятят, чтобы выгнать весь воздух из колбы; затем подводят конец отводной трубки под измерительную трубку, наполненную водой, и приливают в колбу постепенно через воронку раствор анализируемого вещества, напр. селитры, не прекращая нагревания; воронку обмывают соляной кислотой, которую тоже пропускают в колбу. Образующаяся при реакции окись азота собирается в измерительной трубке. Кипячение продолжают до тех пор, пока не выделяется больше ни одного пузырька газа. По объему NО судят о количестве азота обычным образом (см. Газовый анализ). С веществами, не растворимыми в воде, напр. пироксилином, этот прием неудобен. Тогда поступают таким образом: воронку заменяют стеклянной трубкой, идущей до дна колбы, и соединяют ее с прибором для добывания углекислоты. В колбу наливается крепкая соляная кислота, кладется железная соль (вместо FeCl 2 можно взять и железный купорос), и воздух вытесняется из колбы на холоде током угольной кислоты; затем, открыв колбу, бросают в нее быстро навеску анализируемого вещества, снова пропускают СО 2, чтобы удалить следы воздуха, попавшего в колбу при открывании, и затем, прекратив ток углекислоты, начинают кипятить. Окись азота собирают в вышеописанном приборе (фиг. 1). Когда разложение кончилось, остатки NО вытесняют туда же током углекислоты. При этом способе требуется, чтобы разложение вещества начиналось только при нагревании. Для устранения погрешности, происходящей при описанном способе от значительной растворимости окиси азота в воде, и др., П. Рубцов предложил измерять объем не окиси азота, а свободного азота, для чего выделяющуюся NО он пропускает через накаленную стеклянную трубку с металлической медью, для удержания же паров соляной кислоты газы пропускаются предварительно через обратно поставленный на колбе холодильник, небольшое количество крепкого раствора поташа и трубку с кристаллами кислой углекалиевой соли. Об определении азота по Лунге см. Нитрометр.
С. П. Вуколов. Δ.
| "БРОКГАУЗ И ЕФРОН" >> "Н" >> "НИ" >> "НИТ" |
Статья про "Нитрометрия" в словаре Брокгауза и Ефрона была прочитана 1255 раз
| Пицца в сковороде |
| Ананасы на гриле |
TOP 15
- Волос
- Проно
- Степные животные
- Гимнастика
- Индийский океан
- Архитектура
- Сравнение, в литературе
- Манда
- Клитры
- Колесование
- Испарение
- Травоядные животные
- Оплодотворение у pacтений
- Вредные насекомые
- Электризация тел